Want real case studies? 10 seconds to sign up
Join the platform
Need Regulatory Help? Try Our Platform
Post your regulatory questions or request quotations from verified pharmaceutical consultants worldwide. Get matched with experts who specialize in your market.
May 24, 2025
Approximately 5 minutes
The Importance of Adverse Event Reporting in Hong Kong: Our Insights
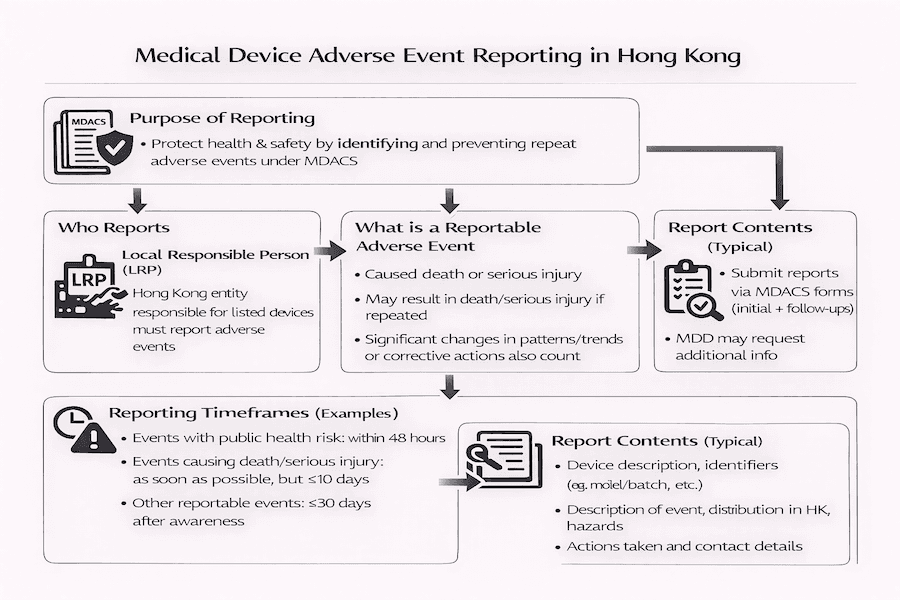
From our perspective, the Medical Device Administrative Control System (MDACS) in Hong Kong places a huge emphasis on keeping an eye on medical devices even after they're sold. This process is called "post-market surveillance," and a crucial part of it is adverse event reporting. The main goal of this reporting system, as explained in guides like GN-03-E, is simple: to make sure everyone's health and safety are better protected. How does it do that? By gathering and sharing information about problems that happen, we can learn from them and stop similar issues with medical devices from happening again. This is vital for medical device safety Hong Kong.
While MDACS is currently an administrative system (meaning it's not a strict law yet), according to our experience, its strong system for reporting adverse events ensures that potential dangers are quickly spotted, investigated, and dealt with. This ultimately keeps both users and patients safe.
Who is Responsible for Reporting? Our Understanding of the Local Responsible Person (LRP) Role
Under the MDACS, the Local Responsible Person (LRP) carries the main burden of reporting and managing any adverse events that happen in Hong Kong involving the medical devices they've listed. From our perspective, this really highlights how important the LRP is – they're the key go-between for the manufacturer, the Medical Device Division (MDD) of the Department of Health, and everyone else involved. Their role is central to regulatory compliance in Hong Kong.
What Exactly is a Reportable Adverse Event? Let's Break It Down.
So, what kind of incident counts as a "reportable adverse event"? Generally, we're talking about any situation involving a medical device that has either already caused, or could potentially cause, certain serious outcomes. While the exact definitions are laid out in official guides, based on our experience, they typically include events that lead to:
- The death of a patient, someone using the device, or another person.
- Serious injury to a patient, user, or another person.
- An event that, if it happened again, might have led to death or serious injury.
This also covers situations where the LRP or manufacturer notices a significant change in how often or in what pattern an issue is occurring – especially if it could lead to serious harm. It also applies when they start taking corrective actions to prevent such harm. These events are key to medical device vigilance.
Reporting Timeframes: Why Speed Matters
Why is it so important to report these events quickly? Because timely reporting allows for swift investigations and appropriate actions to be taken. The MDACS sets specific time limits for telling the MDD about these events. To our understanding, these timeframes usually change depending on how serious the incident is:
- "Expedited reports" (meaning very fast, sometimes within 2 or 10 calendar days) are needed for events that cause death or serious injury, or those that pose a serious threat to public health.
- "Follow-up reports" are required as the investigation progresses, providing more details.
- "Final reports" are submitted once the investigation is complete, giving all the findings and explaining any corrective and preventive actions (CAPA) taken.
The Adverse Event Investigation Process: Our Experience
Once the MDD receives an adverse event report, they might ask the LRP to carry out a very thorough investigation into what happened. The LRP is responsible for this investigation, often working hand-in-hand with the device manufacturer or other relevant parties. The main goal of the investigation is to figure out why the event occurred, identify anything that contributed to it, and put in place the necessary fixes to stop it from happening again. The LRP must then send a detailed report of their findings and recommendations to the MDD. It's also important to remember that the MDD always has the right to conduct its own independent investigation if they feel it's necessary. This ensures accountability in Hong Kong medical device regulation.
Why Comprehensive Documentation and Post-Market Surveillance Are Key
Effective adverse event reporting, from our perspective, heavily relies on solid systems that manufacturers and LRPs have in place for monitoring devices after they've been released to the market. What does this involve?
- Setting up clear, written procedures for identifying, evaluating, and reporting adverse events.
- Keeping thorough records of all complaints, investigations, and corrective actions taken.
- Making sure devices can be easily traced (traceability) so that quick and effective recalls can happen if needed.
According to our experience, staying informed about the very latest guidance notes and regulatory updates from the MDD is absolutely essential for ongoing compliance. This continuous effort significantly contributes to the overall safety and quality of medical devices in the dynamic Hong Kong healthcare market.
Unbeatable Pricing
Transparent, competitive rates for medical device registration in Hong Kong.
ElendiLabs Regulatory Affairs Team
100+ products successfully registered across global markets. Get unbeatable quotations and expert answers — fast.
Ask Anything
We'll follow up with you personally.
100% response rate • Reply within 7 business days
Related Articles
Approximately 5 minutes
Medical Device Adverse Event Reporting in Hong Kong: A Guide for LRPs
Adverse event reporting is a critical component of Hong Kong's Medical Device Administrative Control System (MDACS), aiming to enhance public health and safety. This article outlines the requirements and responsibilities of Local Responsible Persons (LRPs) in reporting adverse events related to listed medical devices, based on our insights.
Approximately 5 minutes
Medical Device Regulations and Registration in Hong Kong
Complete guide to medical device regulations, classification, registration requirements and post-market surveillance in Hong Kong.
Approximately 5 minutes
Navigating Medical Device Regulations in Hong Kong: The MDACS Framework
Hong Kong's Medical Device Administrative Control System (MDACS) provides a robust, although currently voluntary, framework for regulating medical devices. This article explores the system's key features, including device classification, the listing process, the crucial role of Local Responsible Persons (LRPs), and its increasing importance for market access and public procurement, all from our insights and experience.