ElendiLabs
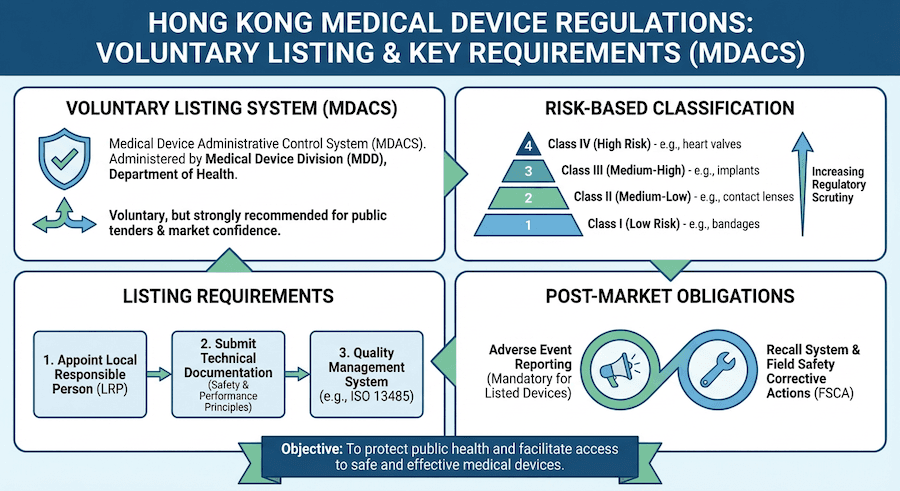
The 2025 revision of TR-007 (Section 5.3.2) clarifies that software which does not directly control hardware but provides information used to take decisions with diagnosis or therapeutic purposes is classified under Rule 12. If your software performs "image analysis for diagnosis," it is generally classified as Class II (at minimum). However, if the analysis relates to critical conditions (e.g., stroke or cancer detection), it may be escalated to Class III. The update emphasizes that "standalone" status no longer offers a pathway to Class I if the clinical impact of the software’s output is high.
Anonymous
Our SaMD (Software as a Medical Device) performs diagnostic image analysis but does not directly control any hardware. Following the September 2025 update to TR-007, how does the MDD now interpret Classification Rule 12 for standalone software that lacks a 'direct medical purpose' in isolation but is intended for use with a specific imaging modality? Does this automatically trigger a Class II listing even if the software is technically 'standalone'?